Bond Dissociation Energy
Calculate the bond dissociation energy (BDE) of ligands attached to the surface of the core. The calculation consists of five distinct steps:
1. Dissociate all combinations of \({n}\) ligands (\(Y\), see
optional.qd.dissociate.lig_count) a nd an atom from the core (\(X\), seeoptional.qd.dissociate.core_atom) within a radius \(r\) from aforementioned core atom (seeoptional.qd.dissociate.lig_core_distandoptional.qd.dissociate.core_core_dist). The dissociated compound has the general structure of \(XY_{n}\).2. Optimize the geometry of \(XY_{n}\) at the first level of theory (\(1\)). Default: ADF MOPAC [1, 2, 3].
3. Calculate the “electronic” contribution to the BDE (\(\Delta E\)) at the first level of theory (\(1\)): ADF MOPAC [1, 2, 3]. This step consists of single point calculations of the complete quantum dot, \(XY_{n}\) and all \(XY_{n}\)-dissociated quantum dots.
4. Calculate the thermalchemical contribution to the BDE (\(\Delta \Delta G\)) at the second level of theory (\(2\)). Default: ADF UFF [4, 5]. This step consists of geometry optimizations and frequency analyses of the same compounds used for step 3.
\(\Delta G_{tot} = \Delta E_{1} + \Delta \Delta G_{2} = \Delta E_{1} + (\Delta G_{2} - \Delta E_{2})\).
Default Settings
optional:
qd:
dissociate:
core_atom: Cd
core_index: null
lig_count: 2
core_core_dist: 5.0 # Ångström
lig_core_dist: 5.0 # Ångström
lig_core_pairs: 1
topology: {}
keep_files: True
job1: AMSJob
s1: True
job2: AMSJob
s2: True
Arguments
- optional.qd.dissociate
optional: qd: dissociate: core_atom: Cd core_index: null lig_count: 2 lig_pairs: 1 core_core_dist: null # Ångström lig_core_dist: 5.0 # Ångström topology: 7: vertice 8: edge 10: face
- optional.qd.dissociate.core_atom
The atomic number or atomic symbol of the core atoms (\(X\)) which are to be dissociated. The core atoms are dissociated in combination with \(n\) ligands (\(Y\), see
dissociate.lig_count). Yields a compound with the general formula \(XY_{n}\).Atomic indices can also be manually specified with
dissociate.core_indexIf one is interested in dissociating ligands in combination with a molecular species (e.g. \(X = {NR_4}^+\)) the atomic number (or symbol) can be substituted for a SMILES string represting a poly-atomic ion (e.g. tetramethyl ammonium: C[N+](C)(C)C).
If a SMILES string is provided it must satisfy the following 2 requirements:
The SMILES string must contain a single charged atom; unpredictable behaviour can occur otherwise.
The provided structure (including its bonds) must be present in the core.
Warning
This argument has no value be default and thus must be provided by the user.
- optional.qd.dissociate.lig_count
- Parameter
Type -
intThe number of ligands, \(n\), which is to be dissociated in combination with a single core atom (\(X\), see
dissociate.core_atom).Yields a compound with the general formula \(XY_{n}\).
Warning
This argument has no value be default and thus must be provided by the user.
- optional.qd.dissociate.core_index
Alternative to
dissociate.lig_core_distanddissociate.core_atom. Manually specify the indices of all to-be dissociated atoms in the core. Core atoms will be dissociated in combination with the \(n\) closest ligands.Note
The yaml format uses
nullrather thanNoneas in Python.
- optional.qd.dissociate.core_core_dist
The maximum to be considered distance (Ångström) between atoms in
dissociate.core_atom. Used for determining the topology of the core atom(see
dissociate.topology) and whether it is exposed to the surface of the core or not. It is recommended to use a radius which encapsulates a single (complete) shell of neighbours.If not specified (or equal to
0.0) CAT will attempt to guess a suitable value based on the cores’ radial distribution function.
- optional.qd.dissociate.lig_core_dist
Dissociate all combinations of a single core atom (see
dissociate.core_atom) and the \(n\) closests ligands within a user-specified radius.Serves as an alternative to
dissociate.lig_core_dist, which removes a set number of combinations rather than everything withing a certain radius.The number of ligands dissociated in combination with a single core atom is controlled by
dissociate.lig_count.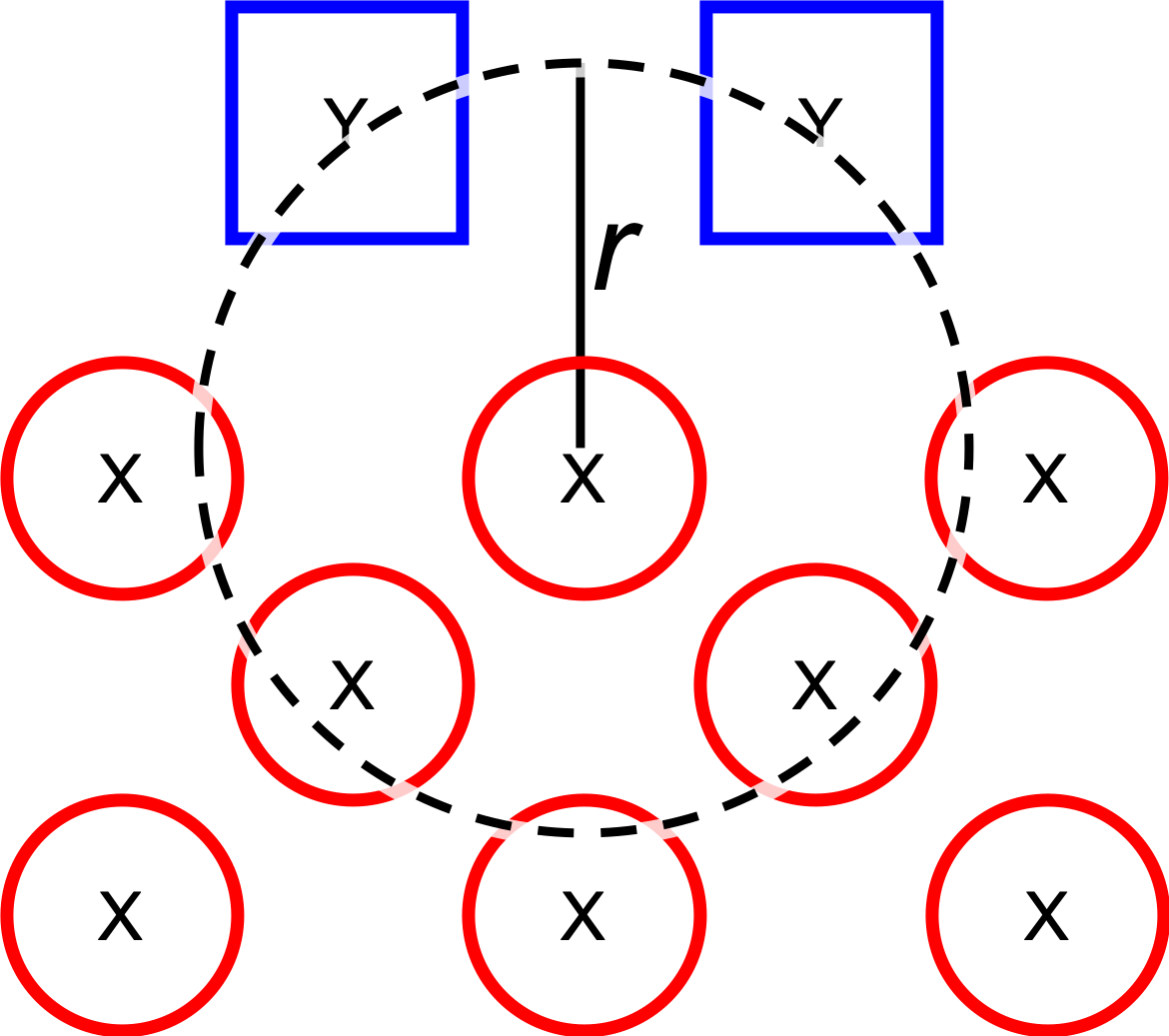
- optional.qd.dissociate.lig_pairs
- Parameter
Type -
int, optionalDefault value –
NoneDissociate a user-specified number of combinations of a single core atom (see
dissociate.core_atom) and the \(n\) closests ligands.Serves as an alternative to
dissociate.lig_core_dist, removing a preset number of (closest) pairs rather than all combinations within a certain radius.The number of ligands dissociated in combination with a single core atom is controlled by
dissociate.lig_count.
- optional.qd.dissociate.topology
- Parameter
Type -
dictDefault value –
{}A dictionary which translates the number neighbouring core atoms (see
dissociate.core_atomanddissociate.core_core_dist) into a topology. Keys represent the number of neighbours, values represent the matching topology.Example
Given a
dissociate.core_core_distof5.0Ångström, the following options can be interpreted as following:optional: qd: dissociate: 7: vertice 8: edge 10: faceCore atoms with
7other neighbouring core atoms (within a radius of5.0Ångström) are marked as"vertice", the ones with8neighbours are marked as"edge"and the ones with10neighbours as"face".
Arguments - Job Customization
- optional.qd.dissociate
optional: qd: dissociate: keep_files: True job1: AMSJob s1: True job2: AMSJob s2: True
- optional.qd.dissociate.keep_files
- Parameter
Type -
boolDefault value –
TrueWhether to keep or delete all BDE files after all calculations are finished.
- optional.qd.dissociate.xyn_pre_opt
- Parameter
Type -
boolDefault value –
TruePre-optimize the \(XY_{n}\) fragment with UFF.
Note
Requires AMS.
- optional.qd.dissociate.job1
A
typeobject of aJobsubclass, used for calculating the “electronic” component (\(\Delta E_{1}\)) of the bond dissociation energy. Involves single point calculations.Alternatively, an alias can be provided for a specific job type (see Type Aliases).
Setting it to
Truewill default toAMSJob, whileFalseis equivalent tooptional.qd.dissociate=False.
- optional.qd.dissociate.s1
The job settings used for calculating the “electronic” component (\(\Delta E_{1}\)) of the bond dissociation energy.
Alternatively, a path can be provided to .json or .yaml file containing the job settings.
Setting it to
Truewill default to the["MOPAC"]block in CAT/data/templates/qd.yaml, whileFalseis equivalent tooptional.qd.dissociate=False.
- optional.qd.dissociate.job2
A
typeobject of aJobsubclass, used for calculating the thermal component (\(\Delta \Delta G_{2}\)) of the bond dissociation energy. Involves a geometry reoptimizations and frequency analyses.Alternatively, an alias can be provided for a specific job type (see Type Aliases).
Setting it to
Truewill default toAMSJob, whileFalsewill skip the thermochemical analysis completely.
- optional.qd.dissociate.s2
The job settings used for calculating the thermal component (\(\Delta \Delta G_{2}\)) of the bond dissociation energy.
Alternatively, a path can be provided to .json or .yaml file containing the job settings.
Setting it to
Truewill default to the the MOPAC block in CAT/data/templates/qd.yaml, whileFalsewill skip the thermochemical analysis completely.
Index
|
Remove \(XY_{n}\) from mol with the help of the |
|
The |
|
Remove out atoms specified in |
Assign a topology to all core atoms in |
|
|
Create and return the indices of each core atom and the \(n\) closest ligands. |
|
Create and return the indices of each core atom and all ligand pairs with max_dist. |
|
Create a list with all to-be removed atom combinations. |
|
Start the dissociation process. |
API
- nanoCAT.bde.dissociate_xyn.dissociate_ligand(mol, lig_count, lig_core_pairs=1, lig_core_dist=None, core_atom=None, core_index=None, core_smiles=None, core_core_dist=None, topology=None, **kwargs)[source]
Remove \(XY_{n}\) from mol with the help of the
MolDissociaterclass.The dissociation process consists of 5 general steps:
Constructing a
MolDissociaterinstance for managing the dissociation workflow.Assigning a topology-descriptor to each atom with
MolDissociater.assign_topology().Identifying all valid core/ligand pairs using either
MolDissociater.get_pairs_closest()orMolDissociater.get_pairs_distance().Creating all to-be dissociated core/ligand combinations with
MolDissociater.get_combinations().Start the dissociation process by calling the earlier created
MolDissociaterinstance.
Examples
>>> from typing import Iterator >>> import numpy as np >>> from scm.plams import Molecule # Define parameters >>> mol = Molecule(...) >>> core_idx = [1, 2, 3, 4, 5] >>> lig_idx = [10, 20, 30, 40] >>> lig_count = 2 # Start the workflow >>> dissociate = MolDissociater(mol, core_idx, lig_count) >>> dissociate.assign_topology() >>> pairs: np.ndarray = dissociate.get_pairs_closest(lig_idx) >>> combinations: Iterator[tuple] = dissociate.get_combinations(pairs) # Create the final iterator >>> mol_iterator: Iterator[Molecule] = dissociate(cor_lig_combinations)
- Parameters
mol (
plams.Molecule) – A molecule.lig_count (
int) – The number of to-be dissociated ligands per core atom/molecule.lig_core_pairs (
int, optional) – The number of to-be dissociated core/ligand pairs per core atom. Core/ligand pairs are picked based on whichever ligands are closest to each core atom. This option is irrelevant if a distance based criterium is used (see lig_dist).lig_core_dist (
float, optional) – Instead of dissociating a given number of core/ligand pairs (see lig_pairs) dissociate all pairs within a given distance from a core atom.core_index (
intorIterable[int]) – An index or set of indices with all to-be dissociated core atoms. See core_atom to define core_idx based on a common atomic symbol/number.core_atom (
intorstr, optional) – An atomic number or symbol used for automatically defining core_idx. Core atoms within the bulk (rather than on the surface) are ignored.core_smiles (
str, optional) – A SMILES string representing molecule containing core_idx. Provide a value here if one wants to disociate an entire molecules from the core and not just atoms.core_core_dist (
float, optional) – A value representing the mean distance between the core atoms in core_idx. IfNone, guess this value based on the radial distribution function of mol (this is generally recomended).topology (
Mapping[int,str], optional) – A mapping neighbouring of atom counts to a user specified topology descriptor (e.g."edge","vertice"or"face").**kwargs (
Any) – For catching excess keyword arguments.
- Returns
A generator yielding new molecules with \(XY_{n}\) removed.
- Return type
- Raises
TypeError – Raised if core_atom and core_idx are both
Noneor lig_core_pairs and lig_core_dist are bothNone.
- class nanoCAT.bde.dissociate_xyn.MolDissociater(mol, core_idx, ligand_count, max_dist=None, topology=None)[source]
The
MolDissociaterclass; serves as an API fordissociate_ligand().- Parameters
mol (
plams.Molecule) – A PLAMS molecule consisting of cores and ligands. SeeMolDissociater.mol.core_idx (
intorIterable[int]) – An iterable with (1-based) atomic indices of all core atoms valid for dissociation. SeeMolDissociater.core_idx.ligand_count (
int) – The number of ligands to-be dissociation with a single atom fromMolDissociater.core_idx. SeeMolDissociater.ligand_count.max_dist (
float, optional) – The maximum distance between core atoms for them to-be considered neighbours. IfNone, this value will be guessed based on the radial distribution function of mol. SeeMolDissociater.ligand_count.topology (
dict[int,str], optional) – A mapping of neighbouring atom counts to a user-specified topology descriptor. SeeMolDissociater.topology.
- mol
A PLAMS molecule consisting of cores and ligands.
- Type
- core_idx
An iterable with (1-based) atomic indices of all core atoms valid for dissociation.
- ligand_count
The number of ligands to-be dissociation with a single atom from
MolDissociater.core_idx.- Type
- max_dist
The maximum distance between core atoms for them to-be considered neighbours. If
None, this value will be guessed based on the radial distribution function ofMolDissociater.mol.- Type
float, optional
- MolDissociater.remove_bulk(max_vec_len=0.5)[source]
Remove out atoms specified in
MolDissociater.core_idxwhich are present in the bulk.The function searches for all neighbouring core atoms within a radius
MolDissociater.max_dist. Vectors are then constructed from the core atom to the mean positioon of its neighbours. Vector lengths close to 0 thus indicate that the core atom is surrounded in a (nearly) spherical pattern, i.e. it’s located in the bulk of the material and not on the surface.Performs in inplace update of
MolDissociater.core_idx.- Parameters
max_vec_len (
float) – The maximum length of an atom vector to-be considered part of the bulk. Atoms producing smaller values are removed fromMolDissociater.core_idx. Units are in Angstroem.
- MolDissociater.assign_topology()[source]
Assign a topology to all core atoms in
MolDissociater.core_idx.The topology descriptor is based on:
The number of neighbours within a radius defined by
MolDissociater.max_dist.The mapping defined in
MolDissociater.topology, which maps the number of neighbours to a user-defined topology description.
If no topology description is available for a particular neighbouring atom count, then a generic
f"{i}_neighbours"descriptor is used (where i is the neighbouring atom count).Performs an inplace update of all
Atom.properties.topologyvalues.
- MolDissociater.get_pairs_closest(lig_idx, n_pairs=1)[source]
Create and return the indices of each core atom and the \(n\) closest ligands.
- Parameters
- Returns
A 2D array with the indices of all valid ligand/core pairs.
- Return type
2D
numpy.ndarray[int]
- MolDissociater.get_pairs_distance(lig_idx, max_dist=5.0)[source]
Create and return the indices of each core atom and all ligand pairs with max_dist.
- MolDissociater.combinations(cor_lig_pairs, lig_mapping=None, core_mapping=None)[source]
Create a list with all to-be removed atom combinations.
- Parameters
cor_lig_pairs (
numpy.ndarray) – An array with the indices of all core/ligand pairs.lig_mapping (
Mapping, optional) – A mapping for translating (1-based) atomic indices incor_lig_pairs[:, 0]to lists of (1-based) atomic indices. Used for mapping ligand anchor atoms to the rest of the to-be dissociated ligands.core_mapping (
Mapping, optional) – A mapping for translating (1-based) atomic indices incor_lig_pairs[:, 1:]to lists of (1-based) atomic indices. Used for mapping core atoms to the to-be dissociated sub structures.
- Returns
A set of 2-tuples. The first element of each tuple is a
frozensetwith the (1-based) indices of all to-be removed core atoms. The second element contains afrozensetwith the (1-based) indices of all to-be removed ligand atoms.- Return type