Bond Dissociation Energy¶
Calculate the bond dissociation energy (BDE) of ligands attached to the surface of the core. The calculation consists of five distinct steps:
1. Dissociate all combinations of n ligands (Y, see qd.dissociate.lig_count) and an atom from the core (X, see qd.dissociate.core_atom) within a radius r from aforementioned core atom (see qd.dissociate.lig_core_dist and qd.dissociate.core_core_dist). The dissociated compound has the general structure of XYn.
2. Optimize the geometry of XYn at the first level of theory (lvl1): ADF MOPAC [1, 2, 3].
3. Calculate the “electronic” contribution to the BDE (dE) at the first level of theory (lvl1): ADF MOPAC [1, 2, 3]. This step consists of single point calculations of the complete quantum dot, XYn and all XYn-dissociated quantum dots.
4. Calculate the thermalchemical contribution to the BDE (ddG) at the second level of theory (lvl2): ADF UFF [4, 5]. This step consists of geometry optimizations and frequency analyses of the same compounds used for step 3.
5. dG = dElvl1 + ddGlvl2 = dElvl1 + ( dGlvl2 - dElvl2 ).
Default Settings¶
optional:
qd:
dissociate:
core_atom: Cd
lig_count: 2
core_core_dist: 5.0
lig_core_dist: 5.0
topology:
7: vertice
8: edge
10: face
job1: AMSJob
s1: True
job2: AMSJob
s2: True
Arguments¶
qd.dissociate.core_atom str or int = Cd
The atomic number or atomic symbol of the core atoms (X) which are to be dissociated. The core atoms are dissociated in combination with n ligands (Y, see qd.dissociate.lig_count). Yields a compound with the general formula XYn.
qd.dissociate.lig_count int = 2
The number of ligands, n, which is to be dissociated in combination with a single core atom (X, see qd.dissociate.core_atom). Yields a compound with the general formula XYn.
qd.dissociate.core_core_dist float = 5.0
The maximum to be considered distance (Ångström) between atoms in qd.dissociate.core_atom. Used for determining the topology of the core atom (see qd.dissociate.topology) and whether it is exposed to the surface of the core or not. It is recommended to use a radius which encapsulates a single (complete) shell of neighbours.
qd.dissociate.lig_core_dist float = 5.0
Dissociate all possible combinations of n ligands and a single core atom (see qd.dissociate.core_atom) within a given radius (Ångström) from aforementioned core atom. The number of ligands dissociated in combination with a single core atom is controlled by qd.dissociate.lig_count.
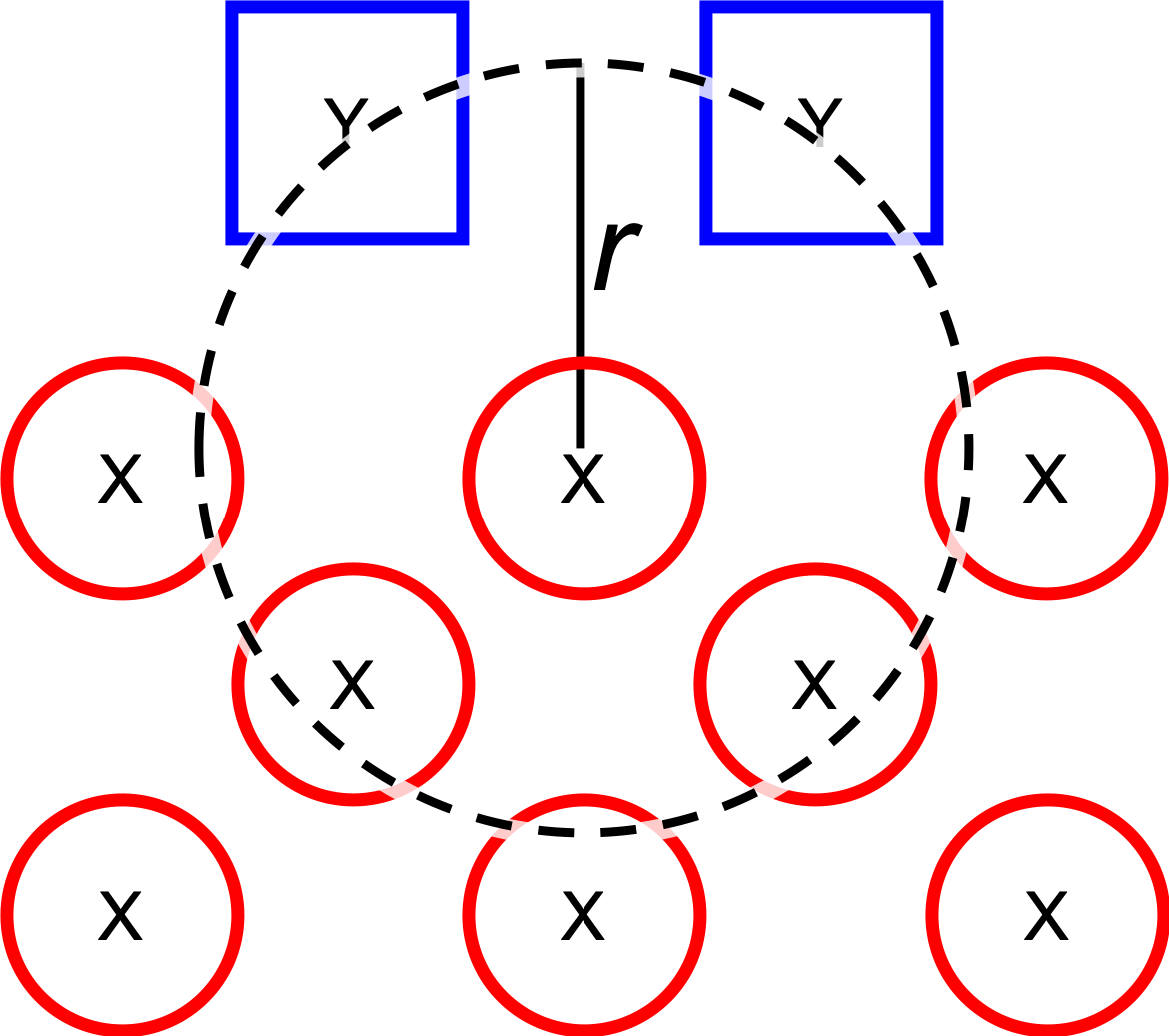
qd.dissociate.topology dict =
{7: vertice, 8: edge, 10: face}
A dictionary which translates the number neighbouring core atoms (see qd.dissociate.core_atom and qd.dissociate.core_core_dist) into a topology. Keys represent the number of neighbours, values represent the matching topology.
Note: values can take on any user-specified value (e.g. Miller indices) and are thus not limited to vertice, edge and/or face.
Arguments - Job Customization¶
qd.dissociate.job1 type, str or bool = AMSJob
A
typeobject of aJobsubclass, used for calculating the “electronic” component (dElvl1) of the bond dissociation energy. Involves single point calculations.Alternatively, an alias (
str) can be provided for a specific job type (see Type Aliases).Setting it to True (
bool) will default totype(AMSJob), while False (bool) is equivalent tooptional.qd.dissociate = False.
qd.dissociate.s1 Settings, str or bool =
s1: input: mopac: model: PM7 ams: system: charge: 0The job
Settingsused for calculating the “electronic” component (dElvl1) of the bond dissociation energy.Alternatively, a path (
str) can be provided to .json or .yaml file containing the job settings.Setting it to True (
bool) will default to the MOPAC block in CAT/data/templates/qd.yaml, while False (bool) is equivalent tooptional.qd.dissociate = False.
qd.dissociate.job2 type, str or bool = AMSJob
A
typeobject of aJobsubclass, used for calculating the thermal component (ddGlvl2) of the bond dissociation energy. Involves a geometry reoptimizations and frequency analyses.Alternatively, an alias (
str) can be provided for a specific job type (see Type Aliases).Setting it to True (
bool) will default totype(AMSJob), while False (bool) will skip the thermochemical analysis completely.
qd.dissociate.s2 Settings, str or bool =
s2: input: uff: library: uff ams: system: charge: 0 bondorders: _1: nullThe job
Settingsused for calculating the thermal component (ddGlvl2) of the bond dissociation energy.Alternatively, a path (
str) can be provided to .json or .yaml file containing the job settings.Setting it to True (
bool) will default to the the MOPAC block in CAT/data/templates/qd.yaml, while False (bool) will skip the thermochemical analysis completely.